Calculus by computer
As we saw in the previous section, almost any time that we want to maximise a likelihood, we need to differentiate it. Very often, merely evaluating a likelihood involves integration (to obtain the marginal distribution of the observable data). Unsurprisingly, there are times when it is useful to perform these tasks by computer. Broadly speaking, numerical integration is computationally costly but stable, while numerical differentiation is cheap but potentially unstable. We will also consider the increasingly important topic of ‘automatic differentiation’.
4.1 Cancellation error
Consider the following R code
a <- 1e16
b <- 1e16 + pi
d <- b - a
d should be , right? Actually
d; pi
## [1] 4
## [1] 3.141593
(d-pi)/pi ## relative error in difference is huge...
## [1] 0.2732395
hence . (d was 4, above, because the computer uses a binary rather than a decimal representation).
As we will see, derivative approximation is an area where we can not possibly ignore cancellation error, but actually it is a pervasive issue. For example, cancellation error is the reason why is not a good basis for a ‘one-pass’ method for calculating . is a simple one pass alternative, and generally much better. It is also a substantial issue in numerical linear algebra. For example, orthogonal matrix decompositions (used for QR, eigen and singular value decompositions), usually involve an apparently arbitrary choice at each successive rotation involved in the decomposition, about whether some element of the result of rotation should be positive or negative: one of the choices almost always involves substantially lower cancellation error than the other, and is the one taken.
Since approximation of derivatives inevitably involves subtracting similar numbers, it is self evident that cancellation error will cause some difficulty. The basic reason why numerical integration is a more stable process (at least for statisticians who usually integrate only positive functions) is because numerical integration is basically about adding things up, rather than differencing them, and this is more stable. e.g.
f <- a + b
(f-2e16-pi)/(2e16+pi) ## relative error in sum, is tiny...
## [1] 4.292037e-17
4.2 Approximate derivatives by finite differencing
Consider differentiating a sufficiently smooth function with respect to the elements of its vector argument . might be something simple like or something complicated like the mean global temperature predicted by an atmospheric GCM, given an atmospheric composition, forcing conditions etc.
A natural way to approximate the derivatives is to use:
where is a small constant and is a vector of the same dimension as , with zeroes for each element except the , which is 1. In a slightly sloppy notation, Taylor’s theorem tells us that
Re-arranging while noting that we have
Now suppose that is an upper bound on the magnitude of , we have
That is to say, we have an upper bound on the finite difference truncation 1 error, assuming that the FD approximation is calculated exactly, without round-off error.
In reality we have to work in finite precision arithmetic and can only calculate to some accuracy. Suppose that we can calculate to one part in (the best we could hope for here is that is the machine precision). Now let be an upper bound on the magnitude of , and denote the computed value of by . We have
and
combining the preceding two inequalities we get
an upper bound on the cancellation error that results from differencing two very similar quantities using finite precision arithmetic. (It should now be clear what the problem was in section 1.1 example 2.)
Now clearly the dependence of the two types of error on is rather different. We want as small as possible to minimise truncation error, and as large as possible to minimise cancellation error. Given that the total error is bounded as follows,
it makes sense to chose to minimise the bound. That is we should choose
So if the typical size of and its second derivatives are similar then
will not be too far from optimal. This is why the square root of the machine precision is often used as the finite difference interval. However, note the assumptions: we require that and to be calculable to a relative accuracy that is a small multiple of the machine precision for such a choice to work well. In reality problems are not always well scaled and a complicated function may have a relative accuracy substantially lower than is suggested by the machine precision. In such cases, see section 8.6 of Gill, Murray and Wright (1981), or section 4.3.3 below.
4.2.1 Other FD formulae
The FD approach considered above is forward differencing. Centred differences are more accurate, but more costly…
…in the well scaled case is about right.
Higher order derivatives can also be useful. For example
which in the well scaled case will be most accurate for .
4.3 Automatic Differentiation: code that differentiates itself
Differentiation of complex models or other expressions is tedious, but ultimately routine. One simply applies the chain rule and some known derivatives, repeatedly. The main problem with doing this is the human error rate and the limits on human patience. Given that differentiation is just the repeated application of the chain rule and known derivatives, couldn’t it be automated? The answer is yes. There are two approaches. The first is symbolic differentiation using a computer algebra package such as Maple or Mathematica. This works, but can produce enormously unwieldy expressions when applied to a complex function. The problem is that the number of terms in a symbolic derivative can be very much larger than for the expression being evaluated. It is also very difficult to apply to a complex simulation model, for example.
The second approach is automatic differentiation. This operates by differentiating a function based directly on the computer code that evaluates the function. There are several different approaches to this, but many use the features of object oriented programming languages to achieve the desired end. The key feature of an object oriented language, from the AD perspective, is that every data structure, or object in such a language has a class and the meaning of operators such as +, -, * etc depends on the class of the objects to which they are applied. Similarly the action of a function depends on the class of its arguments.
Suppose then, that we would like to differentiate
w.r.t. its real arguments , and .
2
In R the code
(x1*x2*sin(x3) + exp(x1*x2))/x3
Now define a new type of object of class "ad" which has a value (a floating point number) and a "grad" attribute. In the current case this "grad" attribute will be a 3-vector containing the derivatives of the value w.r.t. x1, x2 and x3. We can now define versions of the arithmetic operators and mathematical functions which will return class "ad" results with the correct value and "grad" attribute, whenever they are used in an expression.
Here is an R function to create and initialise a simple class "ad" object
ad <- function(x,diff = c(1,1)) {
## create class "ad" object. diff[1] is length of grad
## diff[2] is element of grad to set to 1.
grad <- rep(0,diff[1])
if (diff[2]>0 && diff[2]<=diff[1]) grad[diff[2]] <- 1
attr(x,"grad") <- grad
class(x) <- "ad"
x
}
"grad" attribute, and setting the first element of grad to 1 since .
x1 <- ad(1,c(3,1))
x1
## [1] 1
## attr(,"grad")
## [1] 1 0 0
## attr(,"class")
## [1] "ad"
"ad" objects and correctly propagate derivatives alongside values. Here is a sin function for class "ad".
sin.ad <- function(a) {
grad.a <- attr(a,"grad")
a <- as.numeric(a) ## avoid infinite recursion - change class
d <- sin(a)
attr(d,"grad") <- cos(a) * grad.a ## chain rule
class(d) <- "ad"
d
}
and here is what happens when it is applied to x1
sin(x1)
## [1] 0.841471
## attr(,"grad")
## [1] 0.5403023 0.0000000 0.0000000
## attr(,"class")
## [1] "ad"
"grad" contains the derivative of w.r.t. evaluated at .
Operators can also be overloaded in this way. e.g. here is multiplication operator for class "ad":
"*.ad" <- function(a,b) { ## ad multiplication
grad.a <- attr(a,"grad")
grad.b <- attr(b,"grad")
a <- as.numeric(a)
b <- as.numeric(b)
d <- a*b ## evaluation
attr(d,"grad") <- a * grad.b + b * grad.a ## product rule
class(d) <- "ad"
d
}
Continuing in the same way we can provide a complete library of mathematical functions and operators for the "ad" class. Given such a library, we can obtain the derivatives of a function directly from the code that would simply evaluate it, given ordinary floating point arguments. For example, here is some code evaluating a function.
x1 <- 1; x2 <- 2; x3 <- pi/2
(x1*x2*sin(x3)+ exp(x1*x2))/x3
## [1] 5.977259
"ad" objects
x1 <- ad(1, c(3,1))
x2 <- ad(2, c(3,2))
x3 <- ad(pi/2, c(3,3))
(x1*x2*sin(x3) + exp(x1*x2))/x3
## [1] 5.977259
## attr(,"grad")
## [1] 10.681278 5.340639 -3.805241
## attr(,"class")
## [1] "ad"
This simple propagation of derivatives alongside the evaluation of a function is known as forward mode auto-differentiation. R is not the best language in which to try to do this, and if you need AD it is much better to use existing software libraries in C++, for example, which have done all the function and operator re-writing for you.
4.3.1 Reverse mode AD
If you require many derivatives of a scalar valued function then forward mode AD will have a theoretical computational cost similar to finite differencing, since at least as many operations are required for each derivative as are required for function evaluation. In reality the overheads associated with operator overloading make AD more expensive (alternative strategies also carry overheads). Of course the benefit of AD is higher accuracy, and in many applications the cost is not critical.
An alternative with the potential for big computational savings over finite differencing is reverse mode AD. To understand this it helps to think of the evaluation of a function in terms of a computational graph. Again concentrate on the example function given in Nocedal and Wright (2006):
Any computer evaluating this must break the computation down into a sequence of elementary operations on one or two floating point numbers. This can be thought of as a graph:
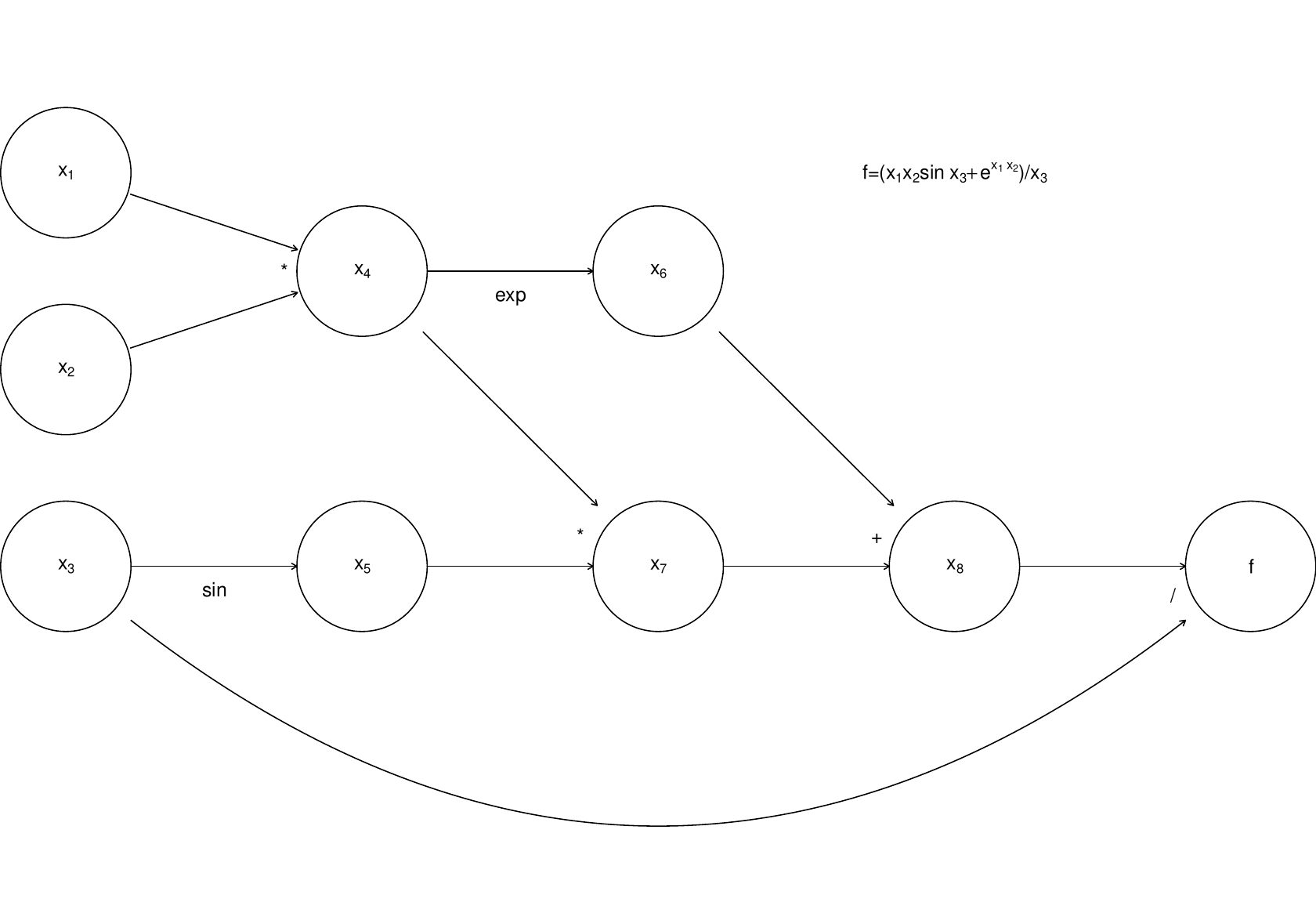
where the nodes to are the intermediate quantities that will have to be produced en route from the input values to to the final answer, . The arrows run from parent nodes to child nodes. No child can be evaluated until all its parents have been evaluated.
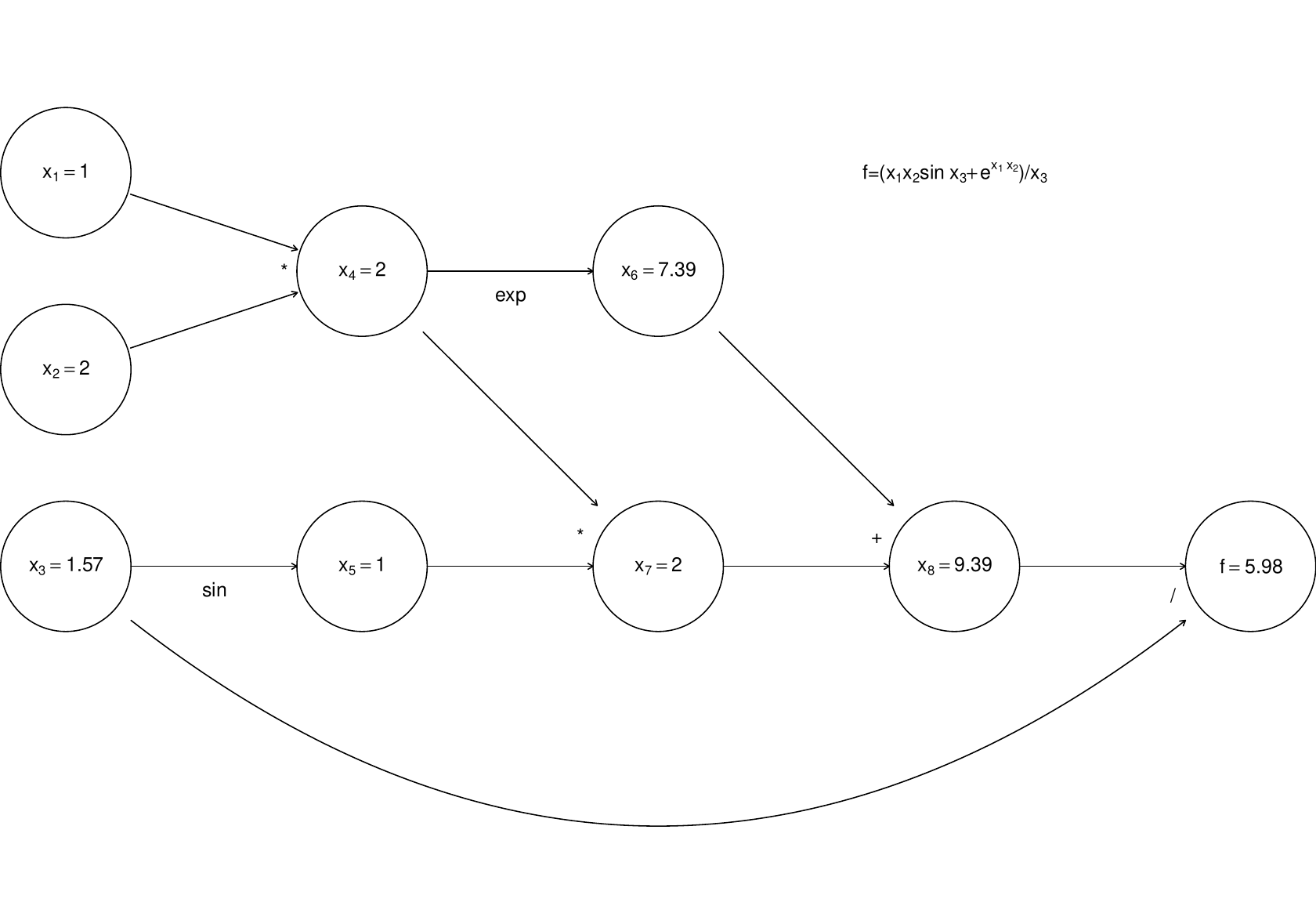
Simple left to right evaluation of this graph results in this …
Now, forward mode AD carries derivatives forward through the graph, alongside values. e.g. the derivative of a node with respect to input variable, is computed using
(the r.h.s. being evaluated by overloaded functions and operators, in the object oriented approach). The following illustrates this process, just for the derivative w.r.t. .
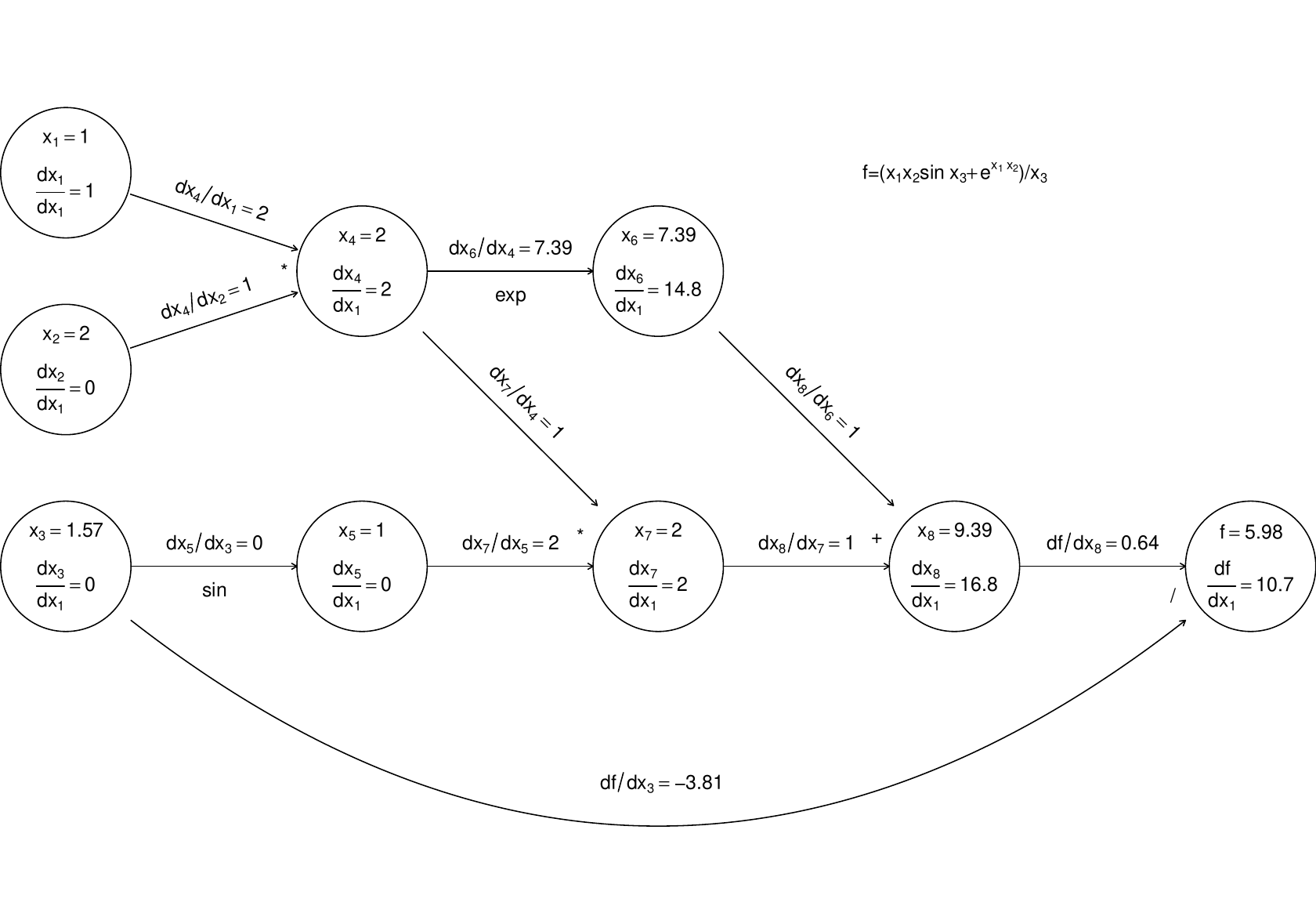
…Again computation runs left to right, with evaluation of a node only possible once all parent values are known.
Now, if we require derivatives w.r.t. several input variables, then each node will have to evaluate derivatives w.r.t. each of these variables, and this becomes expensive (in terms of the previous graph, each node would contain multiple evaluated derivatives). Reverse mode therefore does something ingenious. It first executes a forward sweep through the graph, evaluating the function and all the derivatives of nodes w.r.t. their parents, as follows:
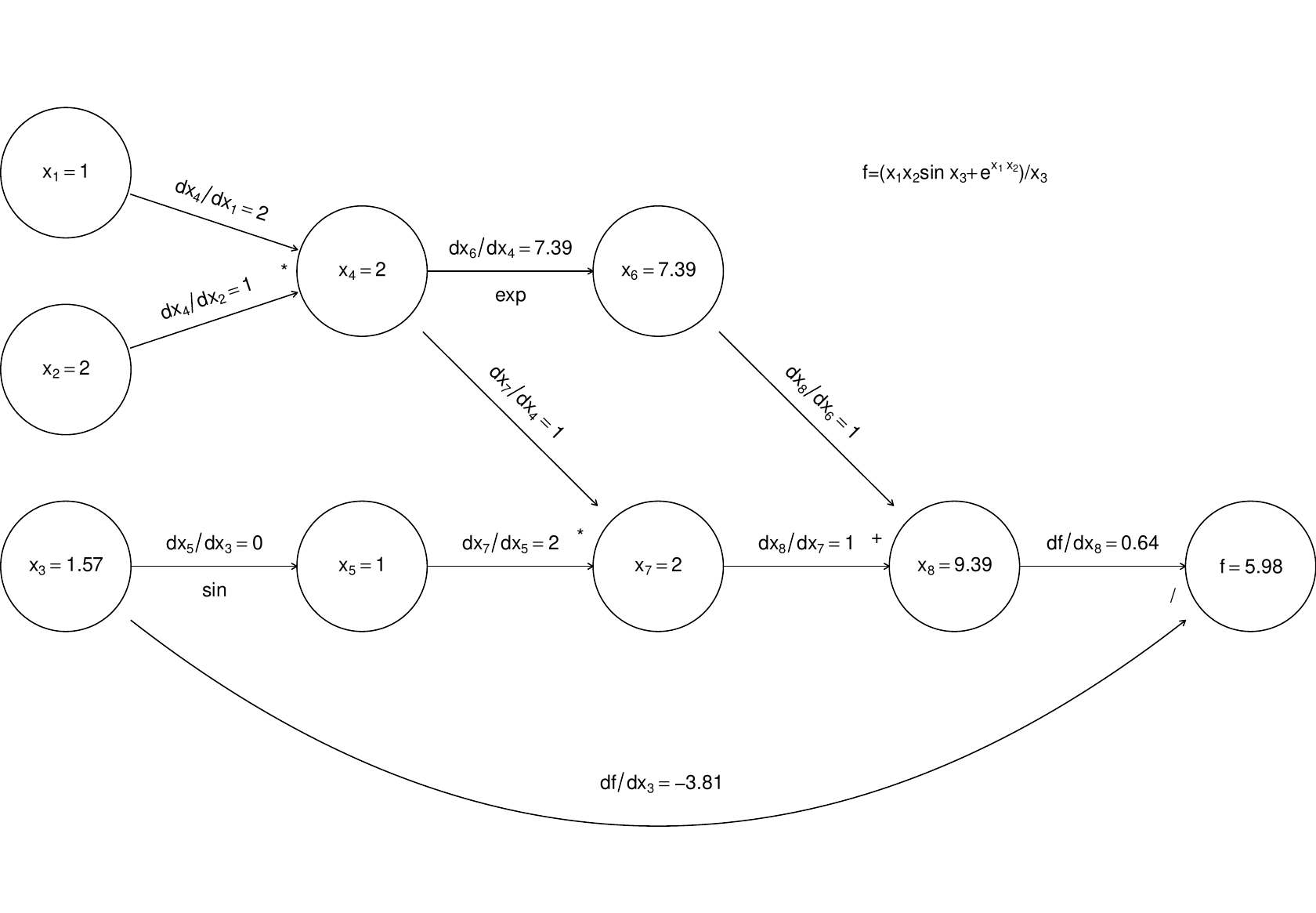
The reverse sweep then works backwards through the graph, evaluating the derivative of w.r.t. each node, using the following
and the fact that at the terminal node .
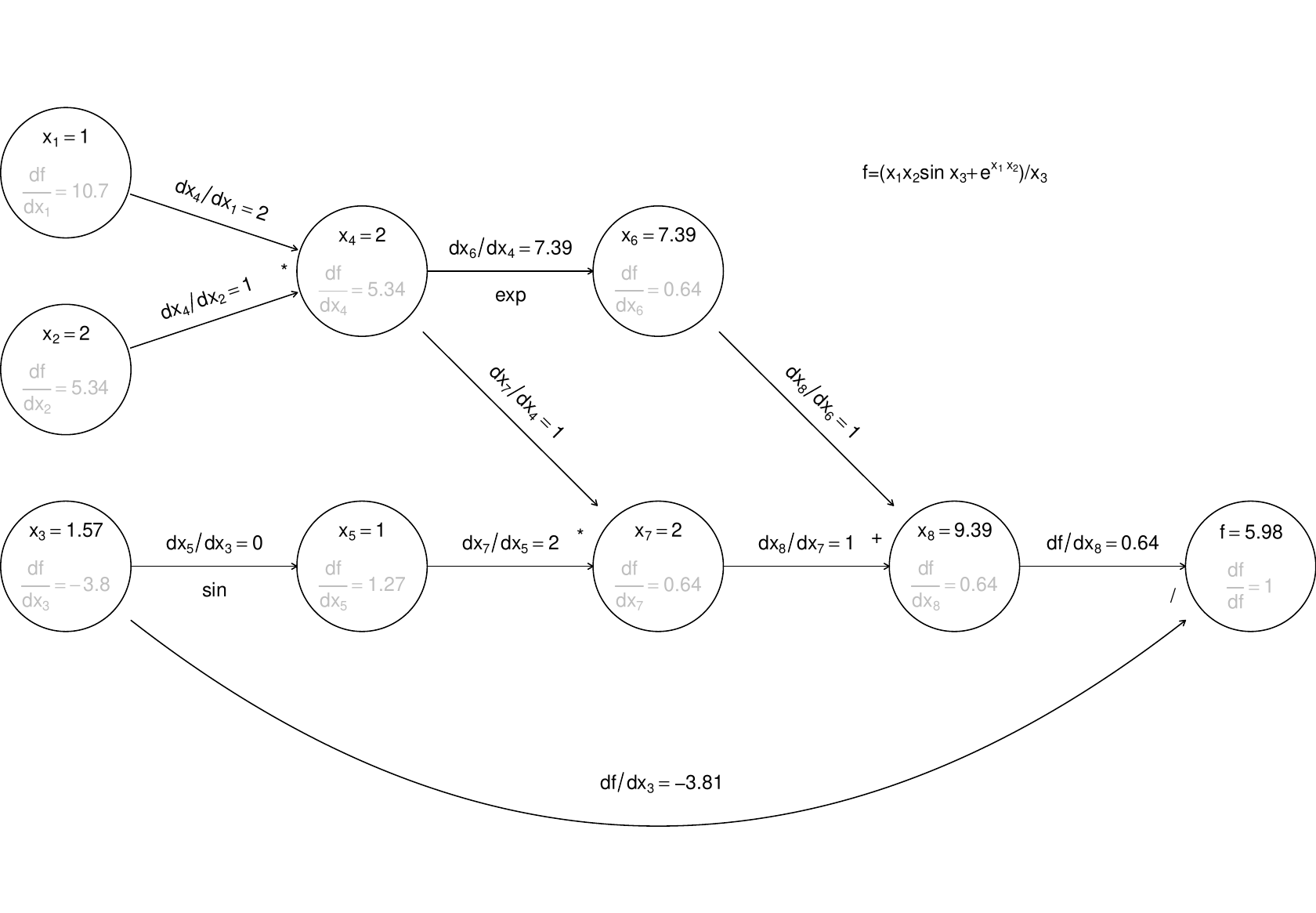
The derivatives in grey are those calculated on the reverse sweep. The point here is that there is only one derivative to be evaluated at each node, but in the end we know the derivative of w.r.t. every input variable. Reverse mode can therefore save a large number of operations relative to finite differencing or forward mode. Once again, general purpose AD libraries automate the process for you, so that all you need to be able to write is the evaluation code.
Unfortunately reverse mode efficiency comes at a heavy price. In forward mode we could discard the values and derivatives associated with a node as soon as all its children were evaluated. In reverse mode the values of all nodes and the evaluated derivatives associated with every connection have to be stored during the forward sweep, in order to be used in the reverse sweep. This is a heavy storage requirement. For example, if involved the inversion of a matrix then we would have to store some intermediate node values plus a similar number of evaluated derivatives. That is some 32 Gigabytes of storage before we even consider the requirements for storing the structure of the graph.
4.3.2 A caveat
For AD to work it is not sufficient that the function being evaluated has properly defined derivatives at the evaluated function value. We require that every function/operator used in the evaluation has properly defined derivatives at its evaluated argument(s). This can create a problem with code that executes conditional on the value of some variable. For example the Box-Cox transformation of a positive datum is
If you code this up in the obvious way then AD will never get the derivative of w.r.t. right if .
4.3.3 Using AD to improve FD
When fitting complicated or computer intensive models it can be that AD is too expensive to use for routine derivative calculation during optimisation. However, with the gradual emergence of highly optimised libraries, this situation is changing, and in any case it can still provide a useful means for calibrating FD intervals. A ‘typical’ model run can be auto-differentiated, and the finite difference intervals adjusted to achieve the closest match to the AD derivatives. As optimisation progresses one or two further calibrations of the FD intervals can be carried out as necessary.
4.4 Numerical Integration
Integration is at least as important as optimisation in statistics. Whether integrating random effects out of a joint distribution to get a likelihood, or just evaluating a simpler expectation, it occurs everywhere. Unfortunately integration is generally numerically expensive, and much statistical research is devoted to clever ways of avoiding it. When it can’t be avoided then the game is typically to find ways of approximating an integral of a function with the minimum number of function evaluations.
-
Integration w.r.t. 1 or 2 (maybe even up to 4) variables is not too problematic. There are two ‘deterministic’ lines of attack.
-
(a)
Use differential equation solving methods since, for example
is the solution of
integrated to .
-
(b)
Quadrature rules. These evaluate the function to be integrated at some set of points in the integration domain and combine the resulting values to obtain an approximation to the integral. For example
is the Midpoint rule for approximate integration. Simpson’s Rule is a higher order variant, while Gaussian Quadrature rules aim for substantially higher order accuracy by careful choice of the locations at which to evaluate.
-
(a)
-
Integration with respect to several-many variables is usually more interesting in statistics and it is difficult. Maintenance of acceptable accuracy with quadrature rules or differential equation methods typically requires an unfeasible number of function evaluations for high dimensional domains of integration. There are two main approaches.
-
(a)
Approximate the function to be integrated by one that can be integrated analytically.
-
(b)
Take a statistical view of the integration process, and construct statistical estimators of the integral. Note that, for a fixed number of function evaluations ,
-
i.
any bias (such as the error in a quadrature rule) scales badly with dimension.
-
ii.
any estimator variance need not depend on dimension at all.
-
iii.
as usual, we can completely eliminate either bias or variance, but not both.
These considerations suggest using unbiased estimators of integrals, based on random samples from the function being integrated. The main challenge is then to design statistical sampling schemes and estimators which minimise the estimator variance.
-
i.
-
(a)
4.4.1 Quadrature rules
In one dimension we might choose to approximate the integral of a continuous function by the area under a set of step functions, with the midpoint of each matching :
where in the final error term is . Let’s see what the error bound means in practice by approximating .
I.true <- exp(1) - 1
N <- 10
I.mp <- sum(exp((1:N-.5)/N))/N
c(I.mp,I.true,(I.true-I.mp)/I.true)
## [1] 1.7175660865 1.7182818285 0.0004165452
N <- 100
I.mp <- sum(exp((1:N-.5)/N))/N
c(I.mp,I.true,(I.true-I.mp)/I.true)
## [1] 1.718275e+00 1.718282e+00 4.166655e-06
N <- 1000
I.mp <- sum(exp((1:N-.5)/N))/N
c(I.mp,I.true,(I.true-I.mp)/I.true)
## [1] 1.718282e+00 1.718282e+00 4.166667e-08
If is cheap to evaluate, accuracy is not paramount and we are only interested in 1 dimensional integrals then we might stop there (or more likely with the slightly more accurate but equally straightforward ‘Simpson’s Rule’), but life is not so simple. If one is very careful about the choice of points at which to evaluate and about the weights used in the integral approximating summation, then some very impressive results can be obtained, at least for smooth enough functions, using Gauss(ian) Quadrature. The basic idea is to find a set such that if is a fixed weight function and is any polynomial up to order then
Note that the set depends on , and , but not on . The hope is that the r.h.s. will still approximate the l.h.s. very accurately if is not a polynomial of the specified type, and there is some impressive error analysis to back this up, for any function with well behaved derivatives (see e.g. Monahan, 2001, section 10.3).
The R package statmod has a nice gauss.quad function for producing the weights, and nodes, , for some standard s. It assumes , , but we can just linearly re-scale our actual interval to use this. Here is the previous example using Gauss Quadrature (assuming is a constant).
library(statmod)
N <- 10
gq <- gauss.quad(N) # get the x_i, w_i
gq # take a look
## $nodes
## [1] -0.9739065 -0.8650634 -0.6794096 -0.4333954 -0.1488743 0.1488743
## [7] 0.4333954 0.6794096 0.8650634 0.9739065
##
## $weights
## [1] 0.06667134 0.14945135 0.21908636 0.26926672 0.29552422 0.29552422
## [7] 0.26926672 0.21908636 0.14945135 0.06667134
sum(gq$weights) # for an interval of width 2
## [1] 2
I.gq <- sum(gq$weights*exp((gq$nodes+1)/2))/2
c(I.gq, I.true, (I.true-I.gq)/I.true)
## [1] 1.718282e+00 1.718282e+00 -1.292248e-15
gqi <- function(g, N = 10, a = -1, b = 1) {
gq = gauss.quad(N)
w = gq$weights*(b-a)/2
n = a + (b-a)*(gq$nodes + 1)/2
sum(w * g(n))
}
gqi(exp, 10, 0, 1)
## [1] 1.718282
I.gq <- gqi(exp, 5, 0, 1)
c(I.gq, I.true, (I.true-I.gq)/I.true)
## [1] 1.718282e+00 1.718282e+00 3.805670e-13
I.gq <- gqi(exp, 3, 0, 1)
c(I.gq, I.true, (I.true-I.gq)/I.true)
## [1] 1.718281e+00 1.718282e+00 4.795992e-07
4.4.2 Quadrature rules for multidimensional integrals
We can integrate w.r.t. several variables by recursive application of quadrature rules. For example consider integrating over a square region, and assume we use basically the same node and weight sequence for both dimensions. We have
and so
Clearly, in principle we can repeat this for as many dimensions as we like. In practice however, to maintain a given level of accuracy, the number of function evaluations will have to rise as where is the number of nodes for each dimension, and is the number of dimensions. Even if we can get away with a 3 point Gaussian quadrature, we will need 1.5 million evaluations by the time we reach a 13 dimensional integral.
Here is a function which takes the node sequence x and weight sequence w of some quadrature rule and uses them to produce the d dimensional set of nodes, and the corresponding weight vector resulting from recursive application of the 1D rule.
mesh <- function(x,d,w=1/length(x)+x*0) {
n <- length(x)
W <- X <- matrix(0,n^d,d)
for (i in 1:d) {
X[,i] <- x;W[,i] <- w
x <- rep(x,rep(n,length(x)))
w <- rep(w,rep(n,length(w)))
}
w <- exp(rowSums(log(W))) ## column product of W gives weights
list(X=X,w=w) ## each row of X gives co-ordinates of a node
}
mesh(c(1,2),3)
## $X
## [,1] [,2] [,3]
## [1,] 1 1 1
## [2,] 2 1 1
## [3,] 1 2 1
## [4,] 2 2 1
## [5,] 1 1 2
## [6,] 2 1 2
## [7,] 1 2 2
## [8,] 2 2 2
##
## $w
## [1] 0.125 0.125 0.125 0.125 0.125 0.125 0.125 0.125
fd <- function(x) {exp(rowSums(x))}
N <- 10; d <- 2; N^d # number of function evaluations
## [1] 100
I.true <- (exp(1)-exp(-1))^d
mmp <- mesh((1:N-.5)/N*2-1,d,rep(2/N,N))
I.mp <- sum(mmp$w*fd(mmp$X))
c(I.mp,I.true,(I.true-I.mp)/I.true)
## [1] 5.506013515 5.524391382 0.003326677
N <- 10; d <- 5; N^d # number of function evaluations
## [1] 1e+05
I.true <- (exp(1)-exp(-1))^d
mmp <- mesh((1:N-.5)/N*2-1,d,rep(2/N,N))
I.mp <- sum(mmp$w*fd(mmp$X))
c(I.mp,I.true,(I.true-I.mp)/I.true)
## [1] 71.136612879 71.731695754 0.008295954
Of course Gauss Quadrature is a better bet
N <- 4; d <- 2; N^d
## [1] 16
I.true <- (exp(1)-exp(-1))^d
gq <- gauss.quad(N)
mgq <- mesh(gq$nodes,d,gq$weights)
I.gq <- sum(mgq$w*fd(mgq$X))
c(I.gq,I.true,(I.true-I.gq)/I.true)
## [1] 5.524390e+00 5.524391e+00 2.511325e-07
N <- 4; d <- 5; N^d
## [1] 1024
I.true <- (exp(1)-exp(-1))^d
gq <- gauss.quad(N)
mgq <- mesh(gq$nodes,d,gq$weights)
I.gq <- sum(mgq$w*fd(mgq$X))
c(I.gq,I.true,(I.true-I.gq)/I.true)
## [1] 7.173165e+01 7.173170e+01 6.278311e-07
4.4.3 Approximating the integrand
Beyond a few dimensions, quadrature rules really run out of steam. In statistical work, where we are often interested in integrating probability density functions, or related quantities, it is sometimes possible to approximate the integrand by a function with a known integral. Here, just one approach will be illustrated: Laplace approximation. The typical application of the method is when we have a joint density and want to evaluate the marginal p.d.f. of ,
For a given let be the value of maximising . Then Taylor’s theorem implies that
where is the Hessian of w.r.t. evaluated at . i.e.
and so
From the properties of normal p.d.f.s we have
so
Notice that the basic ingredients of this approximation are the Hessian of the log integrand w.r.t and the values. Calculating the latter is an optimisation problem, which can usually be solved by a Newton type method, given that we can presumably obtain gradients as well as the Hessian. The highest cost here is the Hessian calculation, which scales with the square of the dimension, at worst, or for cheap integrands the determinant evaluation, which scales as the cube of the dimension. i.e. when it works, this approach is much cheaper than brute force quadrature.
4.4.4 Monte-Carlo integration
Consider integrating over some region of volume . Clearly
where uniform over . Generating uniform random vectors, , over , we have the obvious estimator
which defines basic Monte-Carlo Integration. 3 Clearly is unbiased, and if the are independent then
i.e. the variance scales as , and the standard deviation scales as (it’s ). This is not brilliantly fast convergence to , but compare it to quadrature. If we take the midpoint rule then the (bias) error is where is the node spacing. In dimensions then at best we can arrange , implying that the bias is . i.e. once we get above we would prefer the stochastic convergence rate of Monte-Carlo Integration to the deterministic convergence rate of the midpoint rule.
Of course using Simpson’s rule would buy us a few more dimensions, and for smooth functions the spectacular convergence rates of Gauss quadrature may keep it as the best option up to a rather higher , but the fact remains that its bias is still dependent on , while the sampling error of the Monte Carlo method is dimension independent, so Monte Carlo will win in the end.
4.4.5 Stratified Monte-Carlo Integration
One tweak on basic Monte-Carlo is to divide (a region containing) into equal volumed sub-regions, , and to estimate the integral within each from a single drawn from a uniform distribution over . The estimator has the same form as before, but its variance is now
which is usually lower than previously, because the range of variation of within each is lower than within the whole of .
The underlying idea of trying to obtain the nice properties of uniform random variables, but with more even spacing over the domain of integration, is pushed to the limit by Quasi-Monte-Carlo integration, which uses deterministic space filling sequences of numbers as the basis for integration. For fixed , the bias of such schemes grows much less quickly with dimension than the quadrature rules.
4.4.6 Importance sampling
In statistics an obvious variant on Monte-Carlo integration occurs when the integrand factors into the product of a p.d.f. and another term in which case
where the are observations on r.v.s with p.d.f. . A difficulty with this approach, as with simple MC integration, is that a high proportion of the s often fall in places where makes no, or negligible, contribution to the integral. For example, in Bayesian modelling, it is quite possible for the to be have non-vanishing support over only a tiny volume of the parameter space over which has non-vanishing support.
One approach to this problem is importance sampling. Rather than generate the from we generate from a distribution with the same support as , but with a p.d.f., , which is designed to generate points concentrated in the places where the integrand is non-negligible. Then the estimator is
The expected value of the r.h.s. of the above is its l.h.s., so this estimator is unbiased.
What would make a good ? Suppose that . Then , where . So , and the estimator has zero variance. Clearly this is impractical. If we knew then we would not need to estimate, but there are useful insights here anyway. Firstly, the more constant is , the lower the variance of . Secondly, the idea that the density of evaluation points should reflect the contribution of the integrand in the local neighbourhood is quite generally applicable.
4.4.7 Laplace importance sampling
We are left with the question of how to choose ? When the integrand is some sort of probability density function, a reasonable approach is to approximate it by the normal density implied by the Laplace approximation. That is we take to be the p.d.f. of the multivariate normal where denotes the value maximising , while is the Hessian of w.r.t. , evaluated at .
In fact, as we have seen previously, multivariate normal random vectors are generated by transformation of standard normal random vectors . i.e. where is a Cholesky factor of so that . There is then some computational saving in using a change of variable argument to re-express the importance sampling estimator in terms of the density of , rather than itself. Start with
where is the standard normal density for , giving
and leading in turn to the Laplace importance sampling estimator
Some tweaks may be required.
-
1.
It might be desirable to make a little more diffuse than the Laplace approximation suggests, to avoid excessive importance weights being assigned to values in the tails of the target distribution.
-
2.
If the integration is in order to obtain a likelihood, which is to be maximised, then it is usually best to condition on a set of values, in order to avoid random variability in the approximate likelihood, which will make optimisation difficult.
-
3.
Usually the log of the integrand is evaluated rather than the integrand itself, and it is the log of the integral that is required. To avoid numerical under/overflow problems note that
the r.h.s. will not overflow or underflow when computed in finite precision arithmetic (for reasonable ).
-
4.
As with simple Monte-Carlo integration, some variance reduction is achievable by stratification. The can be obtained by suitable transformation of random vectors from a uniform distribution on . Instead can be partitioned into equal volume parts, with one uniform vector generated in each…
Tweak 3. is an example of a general technique for avoiding numerical underflow and overflow which has many applications in statistical computing. It is therefore worth examining more carefully.
4.4.8 The log–sum–exp trick
In statistical computing, we rarely want to work with raw probabilities and likelihoods, but instead work with their logarithms. Log-likelihoods are much less likely to numerically underflow or overflow than raw likelihoods. For likelihood computations involving iid observations, this is often convenient, since a product of raw likelihood terms becomes a sum of log-likelihood terms. However, sometimes we have to evaluate the sum of raw probabilities or likelihoods. The obvious way to do this would be to exponentiate the logarithms, evaluate the sum, and then take the log of the result to get back to our log scale. However, the whole point to working on the log scale is to avoid numerical underflow and overflow, so we know that exponentiating raw likelihoods is a dangerous operation which is likely to fail in many problems. Can we do the log–sum–exp operation safely? Yes, we can.
Suppose that we have log (unnormalised) weights (typically probabilities or likelihoods), , but we want to compute the (log of the) sum of the raw weights. We can obviously exponentiate the log weights and sum them, but this carries the risk that all of the log weights will underflow (or, possibly, overflow). So, we want to compute
without the risk of underflow. We can do this by first computing and then noting that
We are now safe, since cannot be bigger than zero, and hence no term can overflow. Also, since we know that for some , we know that at least one term will not underflow to zero. So we are guaranteed to get a sensible finite answer in a numerically stable way. Note that exactly the same technique works for computing a sample mean (or other weighted sum) as opposed to just a simple sum (as above).
4.4.9 An example
Consider a Generalised Linear Mixed Model:
where is diagonal and depends on a small number of parameters. Here, are the fixed effects and are the random effects, with covariate matrices and , respectively. From the model specification it is straightforward to write down the joint probability function of and , based on the factorisation . i.e.
but likelihood based inference actually requires that we can evaluate the marginal p.f. for any values of the model parameters. Now
and evaluation of this requires numerical integration.
To tie things down further, suppose that the fixed effects part of the model is simply where is a continuous covariate, while the random effects part is given by two factor variables, and , each with two levels. So a typical row of is a vector of 4 zeros and ones, in some order. Suppose further that the first two elements of relate to and the remainder to and that . The following simulates 50 observations from such a model.
set.seed(0);n <- 50
x <- runif(n)
z1 <- sample(c(0,1),n,replace=TRUE)
z2 <- sample(c(0,1),n,replace=TRUE)
b <- rnorm(4) ## simulated random effects
X <- model.matrix(~x)
Z <- cbind(z1,1-z1,z2,1-z2)
eta <- X%*%c(0,3) + Z%*%b
mu <- exp(eta)
y <- rpois(n, mu)
lf.yb0 <- function(y, b, theta, X, Z, lambda) {
beta <- theta[1:ncol(X)]
theta <- theta[-(1:ncol(X))]
eta <- X%*%beta + Z%*%b
mu <- exp(eta)
lam <- lambda(theta, ncol(Z))
sum(y*eta - mu - lfactorial(y)) - sum(lam*b^2)/2 +
sum(log(lam))/2 - ncol(Z)*log(2*pi)/2
}
theta contains the fixed effect parameters, i.e. and the parameters of . So in this example, theta is of length 4, with the first 2 elements corresponding to and the last two are . lambda is the name of a function for turning the parameters of into its leading diagonal elements, for example
var.func <- function(theta, nb) c(theta[1], theta[1],
theta[2], theta[2])
Quadrature rules
First try brute force application of the midpoint rule. We immediately have the slight problem of the integration domain being infinite, but since the integrand will vanish at some distance from , we can probably safely restrict attention to the domain .
theta <- c(0,3,1,1)
nm <- 10;m.r <- 10
bm <- mesh((1:nm-(nm+1)/2)*m.r/nm,4,rep(m.r/nm,nm))
lf <- lf.yb(y,t(bm$X),theta,X,Z,var.func)
log(sum(bm$w*exp(lf-max(lf)))) + max(lf)
## [1] -116.5303
log(sum(bm$w*exp(lf-max(lf)))) + max(lf)
## [1] -118.6234
On very smooth functions Gauss quadrature rules were a much better bet, so let’s try a naive application of these again.
nm <- 10
gq <- gauss.quad(nm)
w <- gq$weights*m.r/2 ## rescale to domain
x <- gq$nodes*m.r/2
bm <- mesh(x,4,w)
lf <- lf.yb(y,t(bm$X),theta,X,Z,var.func)
log(sum(bm$w*exp(lf-max(lf)))) + max(lf)
## [1] -136.95
log(sum(bm$w*exp(lf-max(lf)))) + max(lf)
## [1] -125.3369
Laplace approximation
To use the Laplace approximation we need to supplement lf.yb with functions glf.yb and hlf.yb which evaluate the gradient vector and Hessian of the w.r.t. . Given these functions we first find by Newton’s method
b <- rep(0,4)
while(TRUE) {
b0 <- b
g <- glf.yb(y, b, theta, X, Z, var.func)
H <- hlf.yb(y, b, theta, X, Z, var.func)
b <- b - solve(H, g)
b <- as.vector(b)
if (sum(abs(b-b0)) < 1e-5*sum(abs(b)) + 1e-4) break
}
lf.yb0(y,b,theta,X,Z,var.func) + 2*log(2*pi) -
0.5*determinant(H,logarithm=TRUE)$modulus
## [1] -116.9646
## attr(,"logarithm")
## [1] TRUE
Monte Carlo
Simple brute force Monte Carlo Integration follows
N <- 100000
bm <- matrix((runif(N*4)-.5)*m.r,4,N)
lf <- lf.yb(y, bm, theta, X, Z, var.func)
log(sum(exp(lf-max(lf)))) + max(lf) - log(N) + log(m.r^4)
## [1] -119.8479
## [1] -119.3469
Intelligent Monte Carlo
Using the original factorisation of the joint likelihood, we can re-write our required marginal likelihood
as the expectation of the complete data likelihood wrt the random effects distribution. This gives us a natural Monte Carlo approach to estimation.
lf.ygb <- function(y, b, theta, X, Z, lambda) {
beta = theta[1:ncol(X)]
eta = Z%*%b + as.vector(X%*%beta)
colSums(y*eta - exp(eta) - lfactorial(y))
}
N = 100000
lam = var.func(theta[3:4])
bm = matrix(rnorm(4*N, 0, sqrt(lam)), nrow=4)
llv = lf.ygb(y, bm, theta, X, Z, var.func)
max(llv) + log(mean(exp(llv - max(llv))))
## [1] -119.2791
## [1] -119.0786
Laplace Importance Sampling
Using Laplace importance sampling allows the construction of an efficient unbiased estimate of the marginal likelihood. It is not strictly necessary here, since the previous Monte Carlo approach is arguably simpler. But it is very useful for more challenging problems.
N <- 10000
R <- chol(-H)
z <- matrix(rnorm(N*4),4,N)
bm <- b + backsolve(R,z)
lf <- lf.yb(y,bm,theta,X,Z,var.func)
zz.2 <- colSums(z*z)/2
lfz <- lf + zz.2
log(sum(exp(lfz - max(lfz)))) + max(lfz) + 2 * log(2*pi) -
sum(log(diag(R))) - log(N)
## [1] -119.3229
## [1] -119.2882
## [1] -119.2774
Conclusions
The example emphasises that integration is computationally expensive, and that multi-dimensional integration is best approached by approximating the integrand with something analytically integrable or by constructing unbiased estimators of the integral. The insight that variance is likely to be minimised by concentrating the ‘design points’ of the estimator on where the integrand has substantial magnitude is strongly supported by this example, and leads rather naturally, beyond the scope of this course, to MCMC methods. Note that in likelihood maximisation settings you might also consider avoiding intractable integration via the EM algorithm (see e.g. Lange, 2010 or Monahan, 2001).
4.5 Further reading on computer integration and differentiation
-
Chapter 8 of Nocedal, J. & S.J. Wright (2006) Numerical Optimization (2nd ed.), Springer, offers a good introduction to finite differencing and AD, and the AD examples above are adapted directly from this source.
-
Gill, P.E. , W. Murray and M.H. Wright (1981) Practical Optimization Academic Press, Section 8.6 offers the treatment of finite differencing that you should read before using it on anything but the most benign problems. This is one of the few references that tells you what to do if your objective is not ‘well scaled’.
-
Griewank, A. (2000) Evaluating Derivatives: Principles and Techniques of Algorithmic Differentiation SIAM, is the reference for Automatic Differentiation. If you want to know exactly how AD code works then this is the place to start. It is very clearly written and should be consulted before using AD on anything seriously computer intensive.
-
http://www.autodiff.org is a great resource for information on AD and AD tools.
-
If you want to see AD in serious action, take a look at e.g. Kalnay, E (2003) Atmospheric Modelling, Data Assimilation and Predictability.
-
R is not the best language for computational statistics, and automatic differentiation is not particularly well supported. There are better automatic differentiation options in Julia and Python. The JAX library for Python is particularly interesting - https://jax.readthedocs.io/
-
Although most widely-used AD libraries are based on object-oriented programming approaches, arguably the most elegant approaches exploit functional programming languages with strong static type systems. See, for example: Elliott, C. (2018) The simple essence of automatic differentiation. Proc. ACM Program. Lang. 2, ICFP, Article 70. DOI: https://doi.org/10.1145/3236765, and the Dex language - https://github.com/google-research/dex-lang
-
Chapter 10 of Monahan, J.F (2001) Numerical Methods of Statistics, offers more information on numerical integration, including much of the detail skipped over here, although it is not always easy to follow, if you don’t already have quite a good idea what is going on.
-
Lange, K (2010) Numerical Analysis for Statisticians, second edition, Springer. Chapter 18 is good on one dimensional quadrature.
-
Ripley, B.D. (1987, 2006) Stochastic Simulation Chapters 5 and section 7.4 are good discussions of stochastic integration and Quasi Monte Carlo integration.
-
Robert, C.P. and G. Casella (1999) Monte Carlo Statistical Methods Chapter 3 (and beyond) goes into much more depth than these notes on stochastic integration.
-
Press et al. (2007) Numerical Recipes (3rd ed), Cambridge, has a nice chapter on numerical integration, but with emphasis on 1D.
- So called because it is the error associated with truncating the Taylor series approximation to the function.
- This example is taken from Nocedal and Wright, (2006).
- Note that in practice, if is a complicated shape, it may be preferable to generate uniformly over a simpler enclosing region , and just set the integrand to zero for points outside itself.